















- Characterized by granular deposits of immunoglobins (IgG) and complement (C3) on the capillary walls that accumulate, causing the glomerular basement membrane to abnormally thicken
- Patients typically present with extremely high levels of proteinuria, edema, hypoalbuminemia, and hyperlipidemia1
- Thickened walls become damaged and leak protein into the urine2
- 75-80% of patients present with antibodies to M-type phospholipase A-2 receptors (PLA2R) located on podocytes
-
Production of the anti-PLA2R antibodies results in primary (idiopathic) MN1
- Primary MN is the most common cause (20-40%) of non-diabetic related idiopathic nephrotic syndrome in Caucasian adults2
- The remaining patients have secondary MN from an autoimmune disease, malignancy, drugs, infection, or non-identified autoantibodies1
- Incidence rate estimated to be about 12 patients/million in the United States
-
Higher male dominance
- 2:1 male-to-female ratio
- Usually diagnosed in men 50-60 years of age
- Most common in Caucasians, followed by people of Asian, African, or Hispanic ancestry
-
Prognosis among patients widely varies and typically follows a rule of thirds:
- 1/3 will undergo spontaneous remission (among patients with absent or low anti-PLA2R antibodies)
- 1/3 will experience end-stage renal disease (ESRD)
- 1/3 will develop non-progressive chronic kidney disease (CKD)2
-
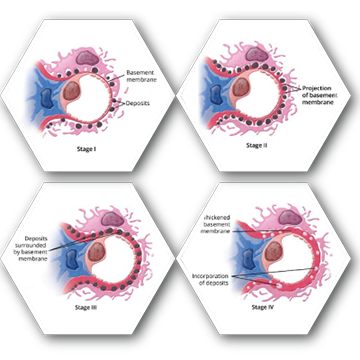
MN gradually progresses and can be identified into a four-stage classification:
- Stage 1 — presence of scattered small immune complex-type deposits on the epithelial side without glomerular basement membrane thickening
- Stage 2 — spiky projections of basement membrane extending between the subepithelial deposits and thickening of the glomerular basement membrane
- Stage 3 — larger deposits surrounded by basement membrane
- Stage 4 — irregular thickening and incorporation of deposits in the basement membrane3
Recommendations are based on the KDIGO Clinical
Practice Guideline for Glomerulonephritis
-
Treatment aimed towards remission of proteinuria to preserve kidney function and ensure patient survival
- Prognosis goals: proteinuria reduction, complete or partial remission of nephrotic syndrome, and stable or improved renal function
- Options are based on degree and persistence of nephrotic-range proteinuria
-
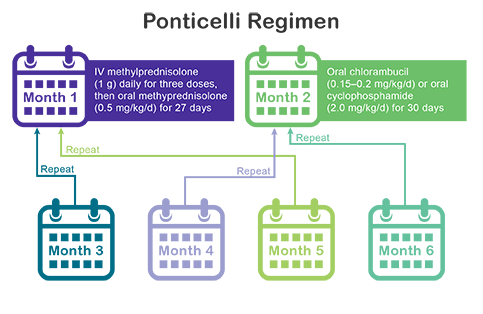
Initial therapy upon diagnosis should consist of alternating monthly cycles of corticosteroids and oral alkylating agents, also known as the Ponticelli Regimen
- KDIGO recommends cyclophosphamide over chlorambucil for the alkylating agent
- At least 6 months of treatment until remission
-
Calcineurin inhibitors (CNIs) are prescribed first line of defense if patients choose not to receive the corticosteroids/alkylating agents therapy or have contraindications
- Options include cyclosporine and tacrolimus
- If a patient is resistant to the corticosteroids/alkylating agents therapy, CNI therapy will be used as an alternative and vice versa
-
As of 2012, the KDIGO guidelines list the following therapies as potential treatment options, but they are not recommended:
- Corticosteroid monotherapy
- Mycophenolate mofetil (MMF)
- Monoclonal antibodies (rituximab)
- Adrenocorticotropic hormone (ACTH)10
Treatment Options
|
|
|---|---|
 Corticosteroids10 Corticosteroids10
|
 Cytotoxic Agents10 Cytotoxic Agents10
|
 Calcineurin Inhibitors10 Calcineurin Inhibitors10
|
 Immunosuppressive Agents10 Immunosuppressive Agents10
|
 Monoclonal Antibodies10 Monoclonal Antibodies10
|
 Acthar® Gel10* Acthar® Gel10*
|
*FDA approved, but not enough contemporaneous data for KDIGO to make a use recommendation